生信学习——生信人的20个R语言习题(下)(附详细答案解读)-程序员宅基地
题目目录
- 12. 理解统计学指标mean,median,max,min,sd,var,mad并计算出每个基因在所有样本的这些统计学指标,最后按照mad值排序,取top 50 mad值的基因,得到列表。
- 13. 根据第12步骤得到top 50 mad值的基因列表来取表达矩阵的子集,并且热图可视化子表达矩阵。试试看其它5种热图的包的不同效果。
- 14. 取不同统计学指标mean,median,max,mean,sd,var,mad的各top50基因列表,使用UpSetR包来看他们之间的overlap情况。
- 15. 在第二步的基础上面提取CLL包里面的data(sCLLex)数据对象的样本的表型数据。
- 16. 添加样本的临床表型数据信息(更改样本名),对所有样本的表达矩阵进行聚类并且绘图。
- 17. 对所有样本的表达矩阵进行PCA分析并且绘图,同样要添加表型信息。
- 18. 根据表达矩阵及样本分组信息进行批量T检验,得到检验结果表格。
- 19. 使用limma包对表达矩阵及样本分组信息进行差异分析,得到差异分析表格,重点看logFC和P值,画个火山图(就是logFC和-log10(P值)的散点图)。
- 20. 对T检验结果的P值和limma包差异分析的P值画散点图,看看哪些基因相差很大。
写在前面——17-20题设计的知识点比较多,本人还没理解透彻,所以没有做完。挖坑挖坑。
(这么多坑,啥时候能填完,/(ㄒoㄒ)/~~)
2021.12.11第一次编辑—— 红色字体
一些新的理解
1. 12-17题中取各种指标进行操作,绘图以及聚类和PCA分析都是为了判断该数据的可用性。聚类的效果越好或PCA分析中不同组分的越开,数据可用性越高。
2. 18,19题都是对数据进行差异分析。提到表达量数据分析,不管是通过芯片技术还是高通量测序技术得到的表达量矩阵,我们都需要根据样本的分组信息来对所检测到的所有基因或者蛋白分子来做差异分析,想找到显著性变化的生物大分子。它们的本质就是对表达量矩阵做一个归一化,让不同组样本的表达量具有可比性,然后利用理想的统计分布检验函数来计算差异的显著性。
3. 更新了19题的代码及注释
题目原文:http://www.bio-info-trainee.com/3409.html
参考答案:https://www.jianshu.com/p/dd4e285665e1 https://www.jianshu.com/p/788010093c90
参考答案:https://www.jianshu.com/p/c62cbb9e1a2e
上篇指路:https://blog.csdn.net/narutodzx/article/details/119775154
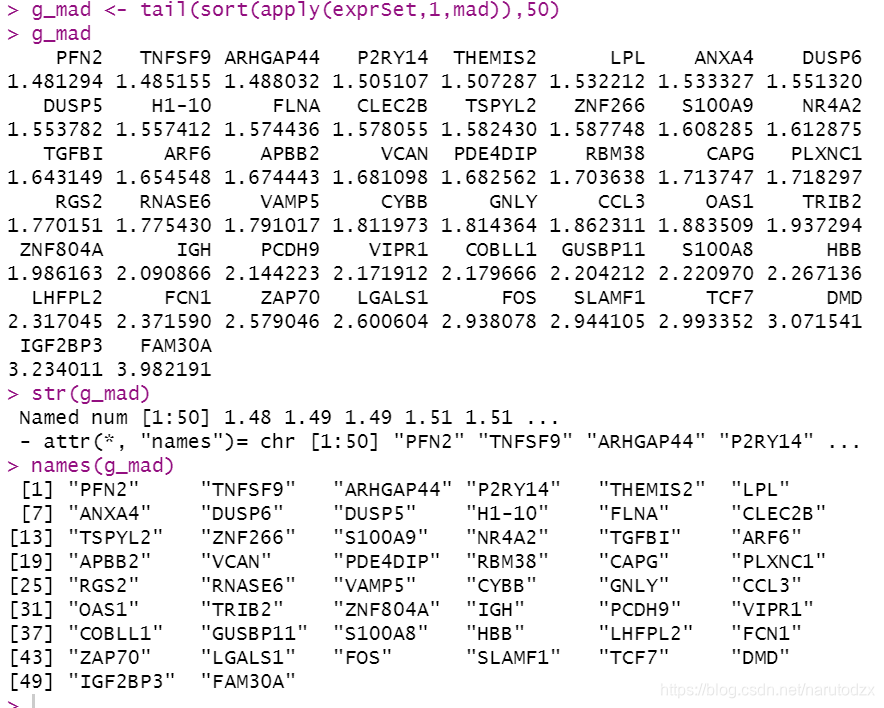
12. 理解统计学指标mean,median,max,min,sd,var,mad并计算出每个基因在所有样本的这些统计学指标,最后按照mad值排序,取top 50 mad值的基因,得到列表。
# 对exprSet的每行求平均值,默认为升序排列,top50即为后50位
g_mean <- tail(sort(apply(exprSet,1,mean)),50)
# 中位数
g_median <- tail(sort(apply(exprSet,1,median)),50)
# 最大值
g_max <- tail(sort(apply(exprSet,1,max)),50)
# 最小值
g_min <- tail(sort(apply(exprSet,1,min)),50)
# 标准差
g_sd <- tail(sort(apply(exprSet,1,sd)),50)
# 方差
g_var <- tail(sort(apply(exprSet,1,var)),50)
# 绝对中位差
g_mad <- tail(sort(apply(exprSet,1,mad)),50)
g_mad
str(g_mad)
names(g_mad)
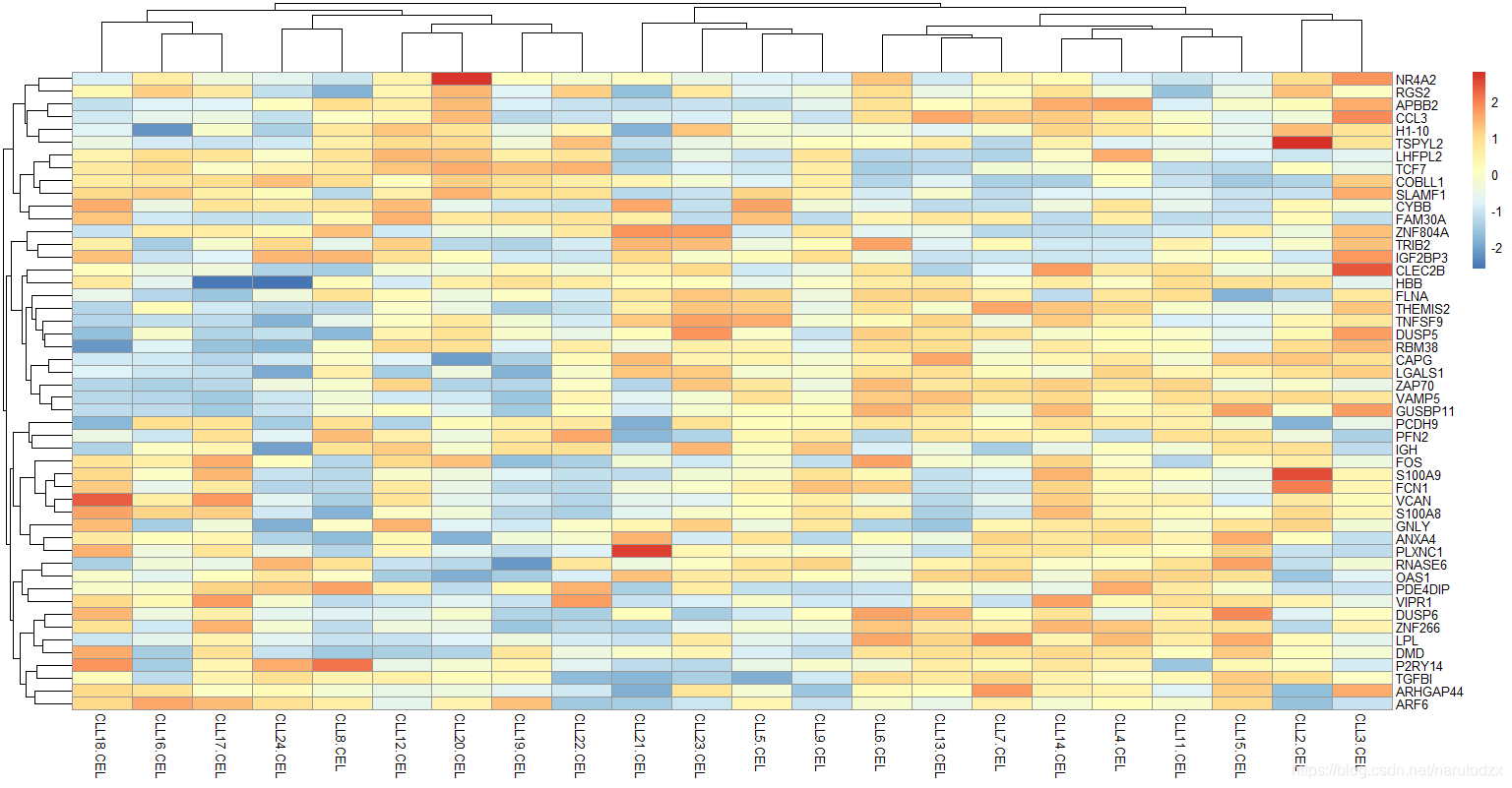
13. 根据第12步骤得到top 50 mad值的基因列表来取表达矩阵的子集,并且热图可视化子表达矩阵。试试看其它5种热图的包的不同效果。
# 紧接上题
library(pheatmap)
# 将top50mad值的基因列表取出
top50mad_exprSet <- exprSet[names(g_mad),]
# scale对数据的“列”进行归一化
# 需要将矩阵转置,scale之后,再转置回来
top50mad_exprSet <- t(scale(t(top50mad_exprSet)))
pheatmap(top50mad_exprSet)
14. 取不同统计学指标mean,median,max,mean,sd,var,mad的各top50基因列表,使用UpSetR包来看他们之间的overlap情况。
# 集合不超过5个时,一般使用韦恩图
# 五个以上可以用UpSetR包进行集合可视化
# install.packages("UpSetR")
library(UpSetR)
# 取这7个统计量的所有基因名,去除重复的
g_all <- unique(c(names(g_mean), names(g_median), names(g_max),
names(g_min), names(g_sd), names(g_var), names(g_mad)))
# 第一列为基因名,第二列是将g_mean在g_all中存在的位置映射为1,反之为0,后面类推
# 最后将所有列合并成数据框
dat <- data.frame(g_all=g_all,
g_mean=ifelse(g_all %in% names(g_mean), 1, 0),
g_median=ifelse(g_all %in% names(g_median), 1, 0),
g_max=ifelse(g_all %in% names(g_max), 1, 0),
g_min=ifelse(g_all %in% names(g_min), 1, 0),
g_sd=ifelse(g_all %in% names(g_sd), 1, 0),
g_var=ifelse(g_all %in% names(g_var), 1, 0),
g_mad=ifelse(g_all %in% names(g_mad), 1, 0)
)
upset(dat, nsets = 7)

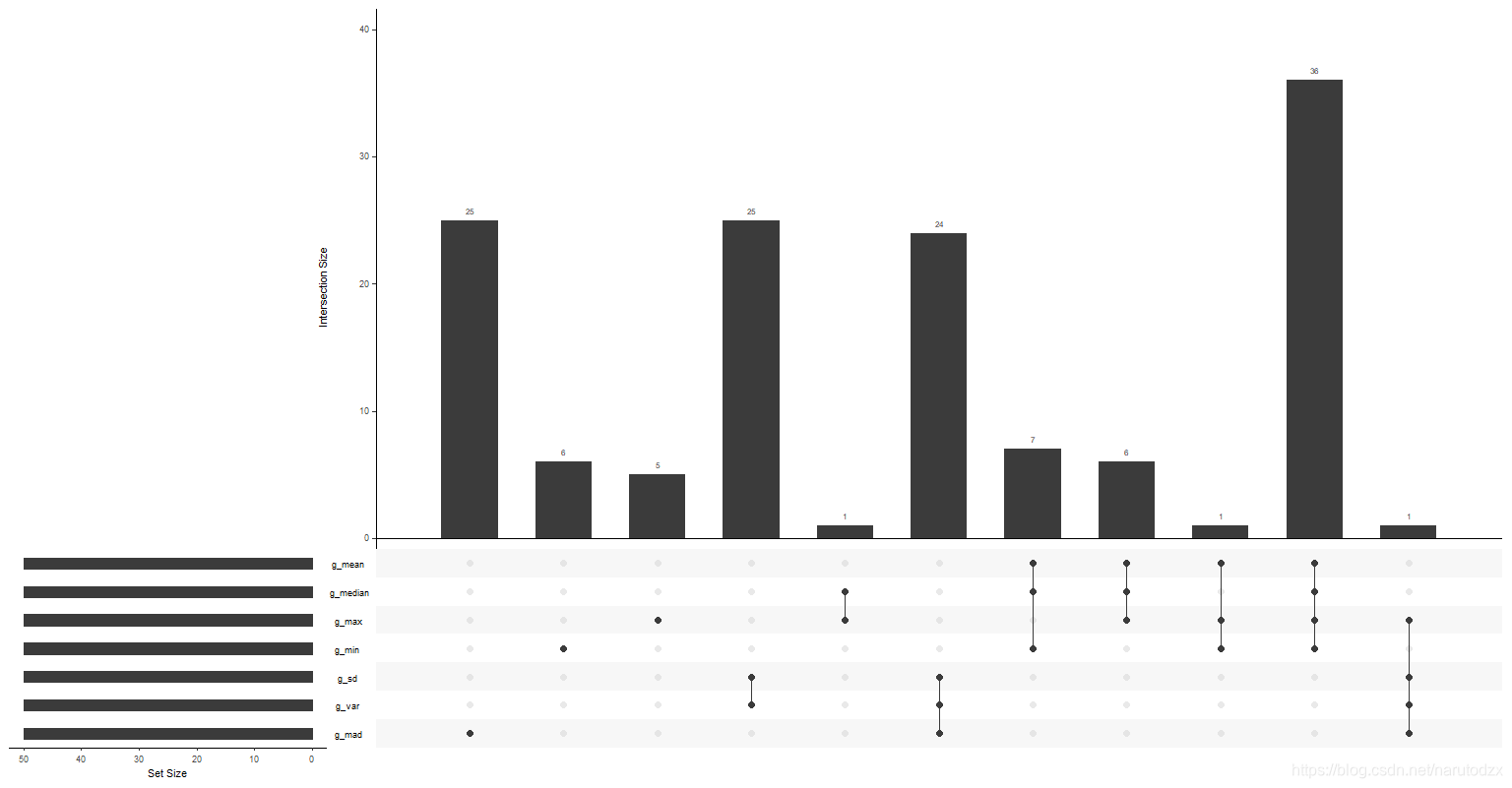
图中左下方:展示了七个集合的名称以及每个集合含多少数据。
图中右侧的直方图和点阵:点的左侧对应集合名称,点阵上方的直方图表示交集数据。
例如:
g_mad这一个点,表示在g_mad中值为1的数量有25个,且这25个基因是g_mad独有的。
g_var和g_sd这两个点连线,表示g_var和g_sd的交集有25个,且这25个基因是g_var和g_sd共同独有的。
把这个想象成韦恩图,会好理解一些…
15. 在第二步的基础上面提取CLL包里面的data(sCLLex)数据对象的样本的表型数据。
pdata <- pData(sCLLex)
group_list <- as.character(pdata[,2])
group_list
table(group_list)

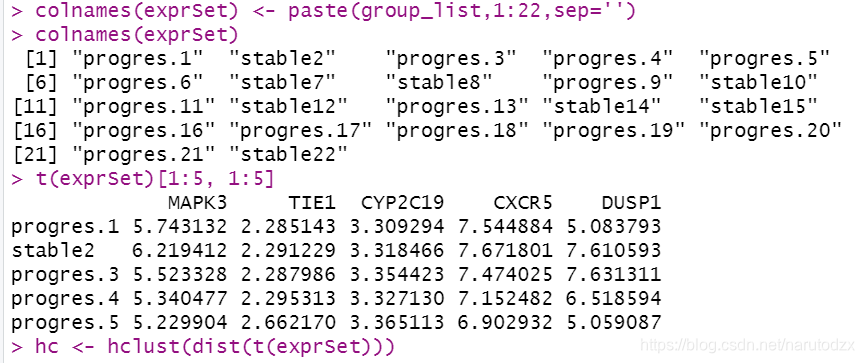
16. 添加样本的临床表型数据信息(更改样本名),对所有样本的表达矩阵进行聚类并且绘图。
# 修改表达矩阵列名,修改为group_list加数字的形式
colnames(exprSet) <- paste(group_list,1:22,sep='')
# dist()计算矩阵所有“行”之间的距离(欧几里得距离)
# 因为要对样本名聚类,所以需要先把exprSet转置
# hclust()来实现层次聚类
hc <- hclust(dist(t(exprSet)))
# 设置图片的各种样式,可以改改数字跑一下,就知道修改的是什么东西了
nodePar <- list(lab.cex = 0.6, pch = c(NA, 19), cex = 0.7, col = "blue")
# 设置plot的四个边指定的边距行数(下,左,上,右)
par(mar=c(5,5,5,10))
# 设置图片为水平,绘图
plot(as.dendrogram(hc), nodePar = nodePar, horiz = TRUE)
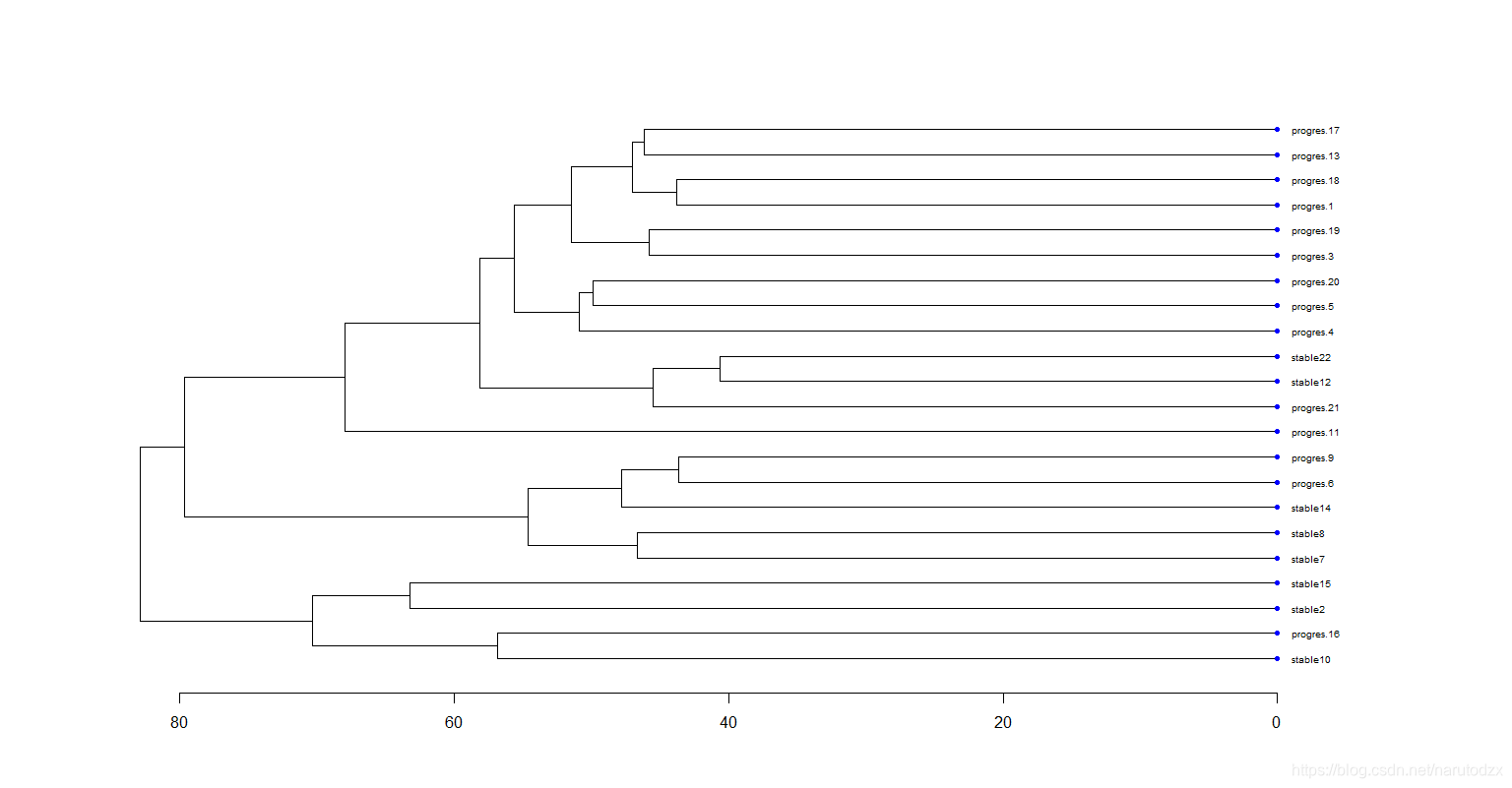
可以看到大部分的progres.或stable都聚集在一起,说明这个数据集是有意义的,可以进行下一阶段的分析。
17. 对所有样本的表达矩阵进行PCA分析并且绘图,同样要添加表型信息。
本题不懂,挖个坑,学透PCA来填。
不懂为啥要重新获得表达矩阵,里面不是有好多与基因不对应的探针嘛,不对应也能当影响指标吗?
转置是因为prcomp只对列操作吗?
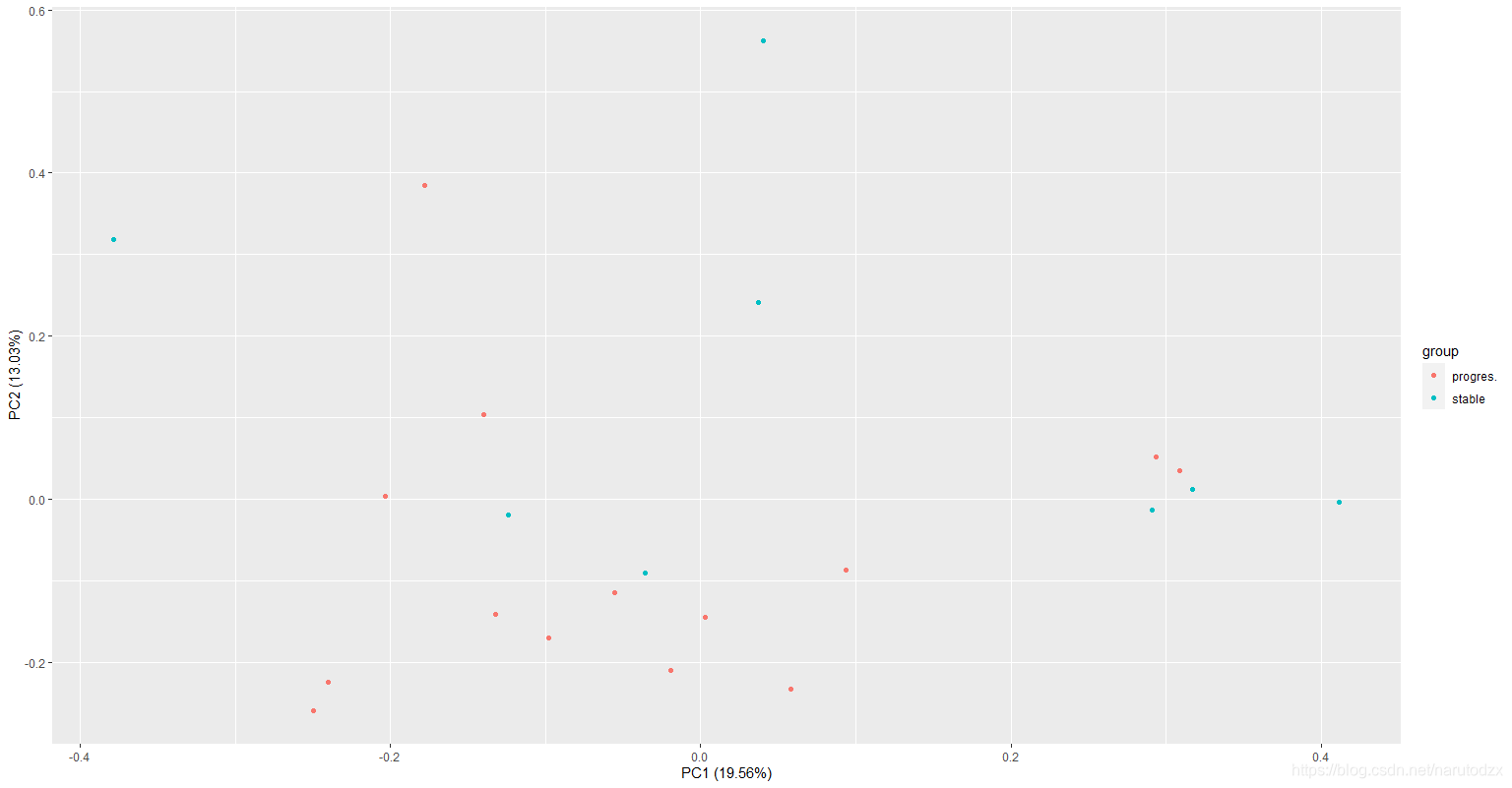
绘制的图片代表什么意思?图片中不同颜色的点分的越开越好,说明我们找的数据可以用
# 主成分分析,也称主分量分析,旨在利用降维的思想,把多指标转化为少数几个综合指标
# install.packages("ggfortify")
library(ggfortify)
exprSet2 <- exprs(sCLLex)
df <- as.data.frame(t(exprSet2))
df$group <- group_list
autoplot(prcomp(df[,1:(ncol(df)-1)]), data=df, colour = 'group')
18. 根据表达矩阵及样本分组信息进行批量T检验,得到检验结果表格。
# 挖坑待埋...
# 注意这时候使用的dat等同于最初的exprSet
dat <- exprs(sCLLex)
# 表型数据因子化
group_list <- as.factor(group_list)
# 整理矩阵
group1 <- which(group_list == levels(group_list)[1])
group2 <- which(group_list == levels(group_list)[2])
dat1 <- dat[, group1]
dat2 <- dat[, group2]
t_dat <- cbind(dat1, dat2)
# T检验,取p.value
pvals <- apply(dat, 1, function(x){
t.test(as.numeric(x) ~ group_list)$p.value
})
# 调整多个比较的p值
p.adj <- p.adjust(pvals, method = "BH")
# log2FC = log2 (mean(处理组/对照组))
# dat是取log2之后的结果
# log2FC = log2(mean(处理组))-log2(mean(对照组)) = avg_2-avg_1
avg_1 <- rowMeans(dat1)
avg_2 <- rowMeans(dat2)
log2FC <- avg_2-avg_1
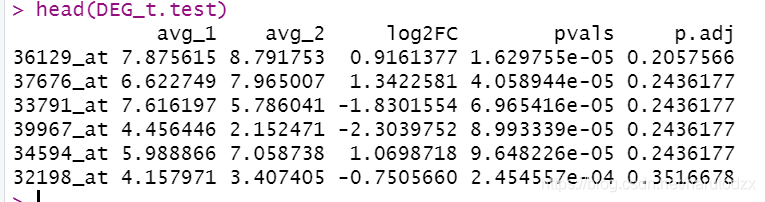
DEG_t.test <- cbind(avg_1, avg_2, log2FC, pvals, p.adj)
DEG_t.test<-DEG_t.test[order(DEG_t.test[,4]),]
DEG_t.test<-as.data.frame(DEG_t.test)
head(DEG_t.test)
19. 使用limma包对表达矩阵及样本分组信息进行差异分析,得到差异分析表格,重点看logFC和P值,画个火山图(就是logFC和-log10(P值)的散点图)。
library(limma)
# 得到按组分离的矩阵
design <- model.matrix(~0 + factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(exprSet)
design
# 差异比较矩阵
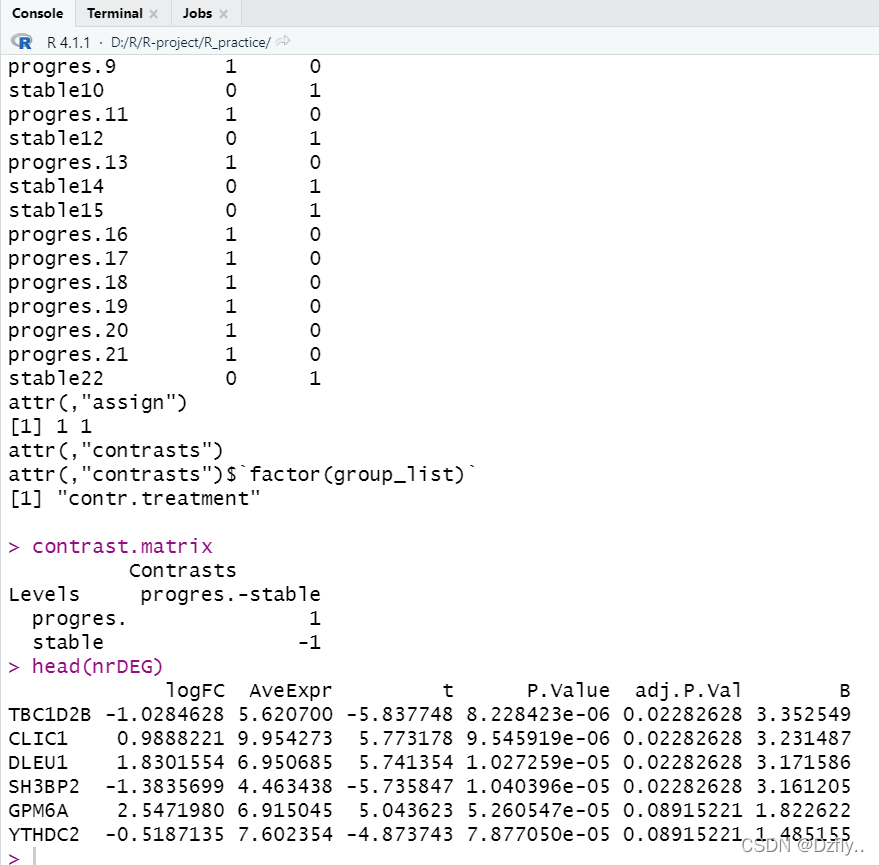
contrast.matrix<-makeContrasts(paste0(unique(group_list),collapse = "-"),levels = design)
contrast.matrix
##step1
# 在给定一系列序列的情况下,对每个基因拟合线性模型
# exprSet要求行对应于基因,列对应于样本
# design要求行对应样本,列对应系数
fit <- lmFit(exprSet,design)
##step2
# 根据lmFit的拟合结果进行统计推断,计算给定一组对比的估计系数和标准误差
# fit由lmFit得到的
# contrasts要求:行对应拟合系数,列包含对比度
fit2 <- contrasts.fit(fit, contrast.matrix)
# Methods of assessing differential expression
fit2 <- eBayes(fit2)
##step3
# 从线性模型拟合中提取出排名靠前的基因表
# For topTable, fit should be an object of class MArrayLM as produced by lmFit and eBayes.
# topTable 默认显示前10个基因的统计数据;使用选项n可以设置,n=Inf就是不设上限,全部输出
# 只有progres.-stable一组的差异基因,就用coef = 1
tempOutput <- topTable(fit2, coef=1, n=Inf)
# 去除缺失值
nrDEG <- na.omit(tempOutput)
head(nrDEG)
## volcano plot
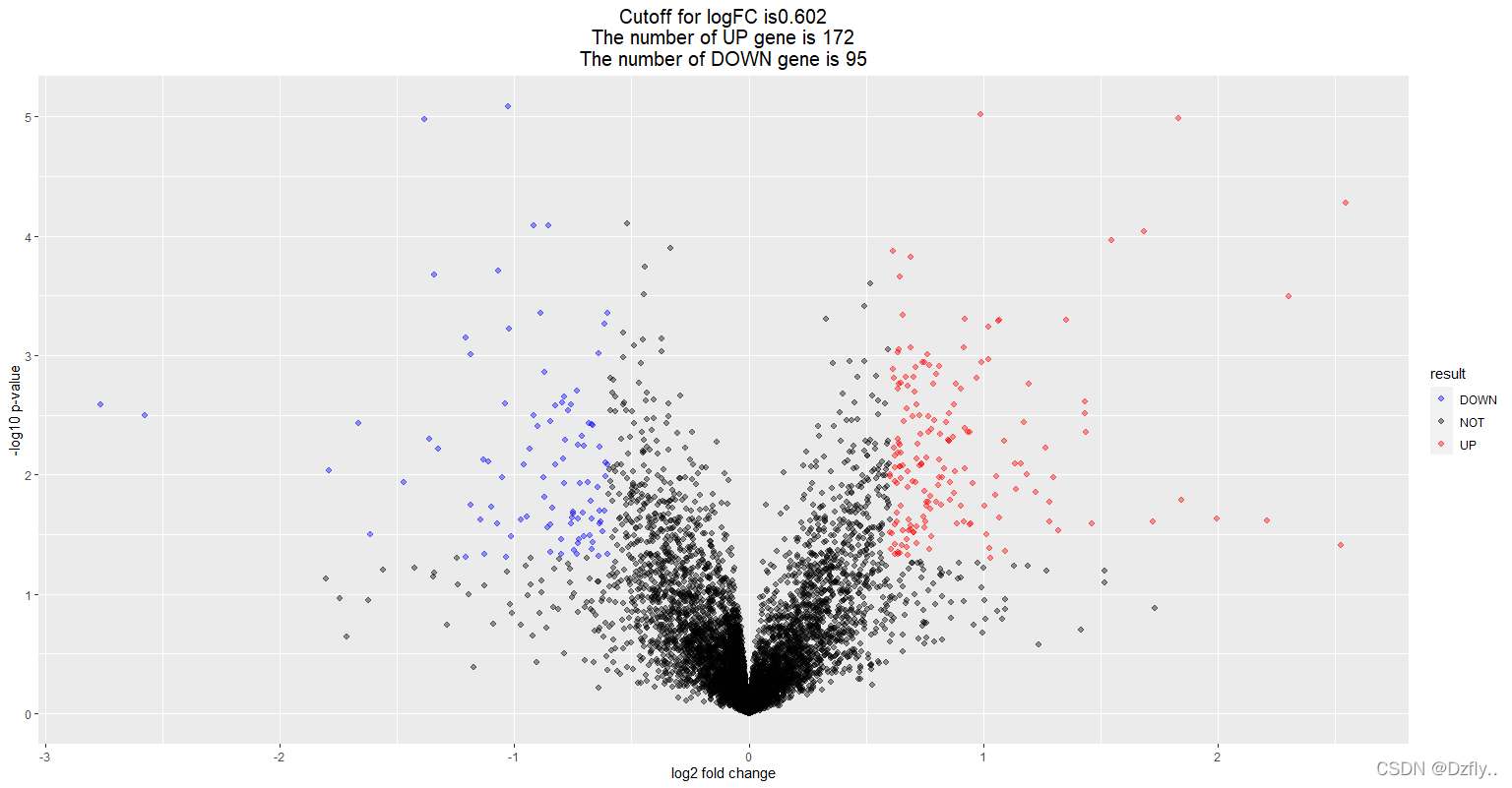
DEG <- nrDEG
# 设定阈值,选出UP、DOWN、NOT表达基因
# mean+2SD可以反映95%以上的观测值,设为mean+3SD,就可以反映97%以上的观测
logFC_cutoff <- with(DEG, mean(abs(logFC)) + 2*sd(abs(logFC)))
# 首先判断p值和logFC的绝对值是不是达到了设定的阈值,如果是则进行下一步判断,如果不是则返回NOT
# 然后判断logFC与阈值的大小关系,返回UP或DOWN
DEG$result <- as.factor(ifelse(DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC >logFC_cutoff, 'UP', 'DOWN'), 'NOT')
)
# 设置火山图标题
this_tile <- paste0('Cutoff for logFC is', round(logFC_cutoff, 3),
'\nThe number of UP gene is ', nrow(DEG[DEG$result == 'UP', ]),
'\nThe number of DOWN gene is ', nrow(DEG[DEG$result == 'DOWN', ]))
this_tile
head(DEG)
library(ggplot2)
# 对p值进行对数转换绘制的图就像火山喷发一样更美观
# 设置一系列的美化条件
ggplot(data=DEG, aes(x=logFC, y=-log10(P.Value), color=result)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red'))
# blue对应DOWN,black对应NOT,red对应UP
20. 对T检验结果的P值和limma包差异分析的P值画散点图,看看哪些基因相差很大。
智能推荐
【ACO TSP】基于matlab蚁群算法求解旅行商问题【含Matlab源码 1583期】-程序员宅基地
文章浏览阅读863次。蚁群算法求解旅行商问题完整的代码,方可运行;可提供运行操作视频!适合小白!
物联网-物联网智能数据处理技术_物联网数据处理技术-程序员宅基地
文章浏览阅读1.9w次,点赞6次,收藏39次。物联网数据处理技术的基本概念物联网数据的特点海量 动态 多态 关联从无线传感器网络TinyDB数据库结构中可以清晰地看到物联网数据“海量、动态、多态、关联”的特点物联网中的数据、信息与知识物联网数据处理关键技术数据存储 数据融合 数据挖掘 智能决策物联网与云计算云计算产生的背景云计算的分类IaaS—基础设施即服务,只涉及到租用硬件,是一种..._物联网数据处理技术
win10找不到打印机_Win10系统如何连接和找寻打印机?-程序员宅基地
文章浏览阅读4.8k次。很多朋友改完win10系统就找不到打印机设备,无法设置默认打印机,今天来解析这个问题!01进入设置界面通常,对于已经启动了并连接到了网络的打印机,会很容易被系统识别到,只不过需要确保打印机和电脑是连接的同一个网络。点击开始菜单,进入设置界面。选择设备。02添加打印机和扫描仪选择打印机和扫描仪,点击添加打印机或扫描仪。系统将会自动搜索识别,并将搜索到的设备罗列出来。接着,找到并点击您想要添加的打印机..._w10打印机在哪里找
【存储缓存】bcache原理及实践-程序员宅基地
文章浏览阅读9.1k次,点赞5次,收藏29次。bcache是linux内核块设备层的cache。主要是使用SSD盘在IO速度较慢的HDD盘上面做一层缓存,从而来提高HDD盘的IO速率。一个缓存设备(SSD)可以同时为多个后端设备(HDD)提供缓存。既然是缓存,那自然就会想到缓存策略,bcache支持三种缓存策略....................._bcache
linux amixer原理,amixer和alsamixer使用说明-程序员宅基地
文章浏览阅读658次。amixer和alsamixer使用说明amixer和alsamixer使用说明amixer和alsamixer说明本文主要解答:1. amixer与alsamixer的区别2. amixer与alsamixer的使用alsamixer与amixer的区别alsamixer是Linux音频框架ALSA工具之一,用于配置音频各个参数;alsamixer是基于文本图形界面的,可以在终端中显示.通过键盘..._amixer
web搭建,dns服务器搭建_dns和web服务器搭建-程序员宅基地
文章浏览阅读2.2k次,点赞3次,收藏15次。安装Web服务1、www(万维网服务),主要应用于搭建web站点2、中间件,是用承载我们的Web站点,那么什么是中间件(如,iis、apache、nginx、tomcat、jboss等),Web网站没有中间件是不能运行。3、如何安装windows IIS服务器管理器–角色–添加–web服务器–4、web站点的访问方式有三种(1)通过ip访问,一般是有多个公网地址,可以每一个站点分配一个ip(这种情况用的极少)原因:Ip很难记,公网地址需要收费(2)多端口访问,web站点默认是80端口,80_dns和web服务器搭建
随便推点
Andorid源码编译需要掌握的shell语法(三)_android shell脚本语法 :>-程序员宅基地
文章浏览阅读1.2k次。Android 源码编译文件中语法记录_android shell脚本语法 :>
Linux V4L2子系统分析(一)_v4l2_subdev_call-程序员宅基地
文章浏览阅读4.2k次,点赞12次,收藏72次。1.概述Linux系统上的Video设备多种多样,如通过Camera Host控制器接口连接的摄像头,通过USB总线连接的摄像头等。为了兼容更多的硬件,Linux内核抽象了V4L2(Video for Linux Two)子系统。V4L2子系统是Linux内核中关于Video(视频)设备的API接口,是V4L(Video for Linux)子系统的升级版本。V4L2子系统向上为虚拟文件系统提供了统一的接口,应用程序可通过虚拟文件系统访问Video设备。V4L2子系统向下给Video设备提供接口,同时管理_v4l2_subdev_call
服务器基础配置:浪潮服务器配置ILO地址、修改管理员密码、查看虚拟化是否打开:_浪潮服务器修改管理口密码-程序员宅基地
文章浏览阅读1w次。使用场景:因为在公司机房中的服务器我们在使用需要对他做一些类似于初始化的配置,分别是三个,——》第一个是配置服务器的ILO地址,这个是我们通过网络打开一个Web页面对服务器进行一些操作;——》第二个是对管理用户的密码进行修改,这个是因为不同的服务器初始的管理员的密码也许是不一样的,我们将其修改为统一的方便记忆也方便管理;——》第三个就是开启服务器的半虚拟化功能,这个是我们的公司的也许需要服..._浪潮服务器修改管理口密码
php如果字符串有1 3 5,PHP常用字符串函数小结-程序员宅基地
文章浏览阅读87次。PHP常用字符串函数小结来源:程序员人生 发布时间:2015-01-22 09:02:32 阅读次数:1594次1、判断类型的函数is_bool() //判断是不是为布尔型is_float() //判断是不是为浮点型is_real() //同上is_int() //判断是不是为整型is_integer() //同上is_string() ..._php 字符串1-5位
matlab从flove,Matlab玩出新高度,变身表白女友神器_善良995的博客-程序员宅基地-程序员宅基地
文章浏览阅读431次。原文作者:善良995原文标题:Matlab玩出新高度,变身表白女友神器发布时间:2021-03-19 13:36:02Matlab还可以这样玩儿?每逢节日愁哭程序员,不知道该送什么给女朋友,在这里教你用Matlab玩儿出属于程序员的浪漫,送给她一整天的惊喜^^一、效果图先来看看效果图:怎么样,这礼物是不是很用心?是不是很特别?是不是很程序猿?(斜眼笑~)二、完整模板代码当然,我怎么忍心让好男孩们千..._clc clear [x,y,z] = meshgrid(linspace(-3,3,101)); f = -x.^2.*z.^3-(9/80)
字符数组和字符串指针在内存中存储_使用字符串指针定义的变量储存在内存中的-程序员宅基地
文章浏览阅读5.5k次,点赞2次,收藏4次。#include#includechar* strcpy1(){ char *p = "hello kitty"; printf("%s\n", p); return p;}int main(){ printf("%s", strcpy1()); return 0;}字符串在内存中存储在只读数据段,当定义一个字符串指针时,该指针指向这个只读区域,即使在函数中将这个指针返回_使用字符串指针定义的变量储存在内存中的