用R分析微生物组群落数据(1) 软件安装、数据集下载和导入_r语言与微生物组学-程序员宅基地
技术标签: 生物信息学 R 数据科学 R语言 bioinformatics readr taxonomy microbiome
参考: https://grunwaldlab.github.io/analysis_of_microbiome_community_data_in_r/index.html
1.下载需要的软件和数据
1.1 安装R、Rstudio和必要的R包
1.1.1 安装R
R是一门关注统计学、数据科学、可视化的编程语言。它可以再所有的公共操作系统安装,R语言可以从如下地址下载:
https://cran.r-project.org/
1.1.2 安装RStudio
RStudio是一个交互式的开发环境( “Integrated Development Environment” ,IDE),即“带有额外编程相关工具的文本编辑器”。RStudio使得R用起来更简单,但是不依赖于R。大多数R语言用户都会使用RStudio, 因此我们推荐使用这个作为工作界面。
RStudio的主要版本是免费开源的,可以通过如下地址下载:
https://www.rstudio.com/products/rstudio/download/#download
1.1.3 安装必要的R包
除了R语言安装过程中包含的包,安装R包是很普遍的。 R语言包是以标准化方式打包到一起的工具的集合,很容易安装。实际上,R语言中的大多数功能是来自于R包,而非基础的R。下面的包将用在这个基础教程中。点击它们的名字就可以跳转到这个包的文档页面:
- vegan: 包括很多能够用于生态学研究的标准化统计工具。被
phyloseq和metacoder等包广泛使用。(Dixon 2003) - metacoder: 处理和可视化分类(taxonomic)数据,特别是来自扩增子宏基因组研究的。(Foster, Sharpton, and Grünwald 2017)
- taxa:定义分类 classes 以及操纵它们的函数。目标是用这些类型作为R包能够构建和使用的低水平基础分类类型。这个能够被
metacoder使用。 (Foster, Chamberlain, and Grünwald 2018) - phyloseq: 包含分析和可视化微生物组数据工具的流行包。 (McMurdie and Holmes 2013)
- ggplot2:漂亮的画图包。(Wickham 2009)
- dplyr:通过内聚和直观的命令集来操纵扁平数据的包。是基本R语言方法的一个流行的替代平。 (Wickham and Francois 2015)
- readr:使得从文件读取扁平数据更加简单。
- stringr:文本操纵的相关函数。
- agricolae:用于实验的设计和分析,特别是植物相关的实验。
- ape :用于DNA序列分析和进化的流行包。
我们写和活跃地维护了metacoder(Foster, Sharpton, and Grünwald 2017) 和taxa(Foster, Chamberlain, and Grünwald 2018), 因此他们非常依赖于这个基本教程。
更多用于微生物组数据分析的R资源可以通过如下链接获取:
https://microsud.github.io/Tools-Microbiome-Analysis/
你可以在R语言编译器中输入下面的代码来安装这些包:
# Install phyloseq from Bioconductor
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install()
BiocManager::install("phyloseq")
# Install the rest of the packages from CRAN
install.packages(c("vegan", "metacoder", "taxa", "ggplot2", "dplyr", "readr", "stringr", "agricolae", "ape"),
repos = "http://cran.rstudio.com",
dependencies = TRUE)
如果安装不成功,可以尝试安装内次安装一个包(如:install.packages("taxa")),然后查找错误信息。通常问题是需要安装一个非R依赖项,如何安装它取决于你的操作系统。将错误信息复制到Google通常可以帮你找出错误原因。如果一个包已经被正确安装,你应该能够用library来加载它(如:library(phyloseq))。
如果你想检测通过开源的Rstudio检查软件是否安装成功,运行下面的代码。如果你使用R或RStudio需要帮助,查看附件:
library(metacoder)
x = parse_tax_data(hmp_otus, class_cols = "lineage", class_sep = ";",
class_key = c(tax_rank = "info", tax_name = "taxon_name"),
class_regex = "^(.+)__(.+)$")
heat_tree(x, node_label = taxon_names, node_size = n_obs, node_color = n_obs)
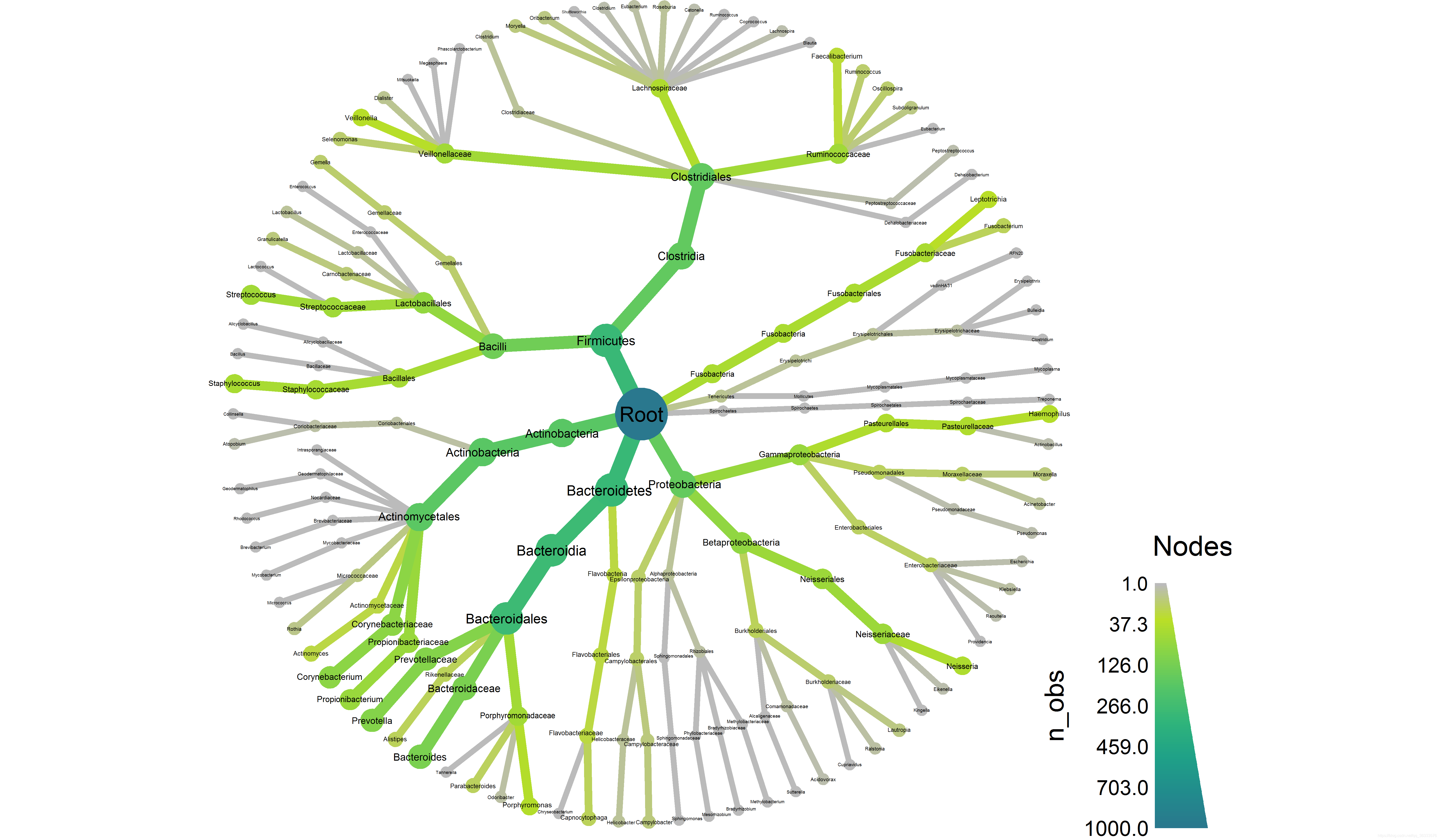
如果你看到了上面的图片,那么祝贺你。你现在应该已经获得这个工作所需的使用R的所有集合。如果你不能够看到这个图,那么请将下面代码运行的结果发邮件给我们,帮助我们排除这个问题的故障。
sessionInfo()
2. 本workshop使用的数据集
在这个workshop中,我们会使来自Wagner et al. (2016)的数据,一个探索植物年龄、基因型、环境对严格布氏小蠊(Boechera stricta,芥菜科多年生草本植物)的细菌群落的影响的研究。Wagner et al. (2016)发布了这篇文章的原始数据,它可以再在 dryad的此处获取。这是一个分享你的原始数据的好例子。
这个数据的副本已经包含在这个网站,能够通过下面的链接下载。
2.1 Sample metadata
sample data是一个表格,每一行是一个样品,样品的信息在每一列,如:植物的基因型和产地。它是一个170kb的以tab分割的文本文件。
SMD.txt
2.2 The OTU abundance table
这个表有每个样品的每个OTU的reads数目。它是一个6Mb压缩的以tab分割的文本文件。
otuTable97.txt.bz2
2.3 The OTU taxonomy file
这个文件有每个OTU的taxonomic classifications,它是一个6Mb的tab键分割的文本文件。
taxAssignments97.txt
3. 将数据输入R
3.1 典型的微生物组数据集
大多数处理高通量amplicon数据的pipeline,例如mothur, QIIME和dada2, 都会产生一个read数目的矩阵。矩阵的一个维度(行或者列)由 Operational Taxonomic Units (OTUs), phylotypes, or exact sequence variants (ESVs)(所有都是二进制的相似读段序列) 组成。另一维由samples组成。不同的工具将期望或者输出不同方向的矩阵,但是在我们的案例中,列是samples,行是OTUs。有时候OTU数据矩阵丰度是两个分开的表格。通常有另外一个行是样本信息的表格。这使得增加很多额外的可作为子集数据的样本数据列更加简单。每一个样本和OTU都有独一无二的ID。

3.2 将数据导入R
如果数据已经格式化得很好了,导入数据到R是很简单的,否则可能是一个令人沮丧的过程。 .csv (comma-separated value)或.tsv (tab-separated value)是格式化好的数据,每一个都是一个单独的表格,没有额外的评论或者形式(如:合并的单元格)。这些格式化的文件都可能有.txt后缀(后缀不是真正重要的,它是针对人的,不是针对计算机的)。正确的数据格式化的更多信息,可以看我们的重现研究指南的数据格式化小节。只要可能,您应该始终导入原始输出数据,并避免任何“手动”操作(如:non-scripted)去修改数据,特别是Excel程序,可能会不时地篡改数据 (Zeeberg et al. (2004))。
在这个workshop中,我们将使用来自Wagner et al. (2016)的数据,一个探索植物年龄、基因型、环境对严格布氏小蠊(Boechera stricta,芥菜科多年生草本植物)的细菌群落的影响的研究。这里是由Mary Ellen Harte拍摄的Boechera stricta的照片:

Wagner et al. (2016)发布了这篇文章的原始数据,它可以再在 dryad的此处获取。这是一个分享你的原始数据的好例子。
有很多functions可以用来读取扁平化数据,包括base R的方法,如read.table和read.csv,但是我们将使用readr package中的函数,其返回tibbles而不是data.frame(R语言中的“table”)。
library(readr) # Loads the readr package so we can use `read_tsv`
Tibbles是一种具有更优美输出和更一致表现的data.frame。点击此处下载OTU表。让我们先读取原始的OTU表:
otu_data <- read_tsv("data/otuTable97.txt.bz2") # You might need to change the path to the file
print(otu_data) # You can also enter just `otu_data` to print it
结果如下:
## # A tibble: 47,806 x 1,699
## OTU_ID M1024P1833 M1551P81 M1551P57 M1551P85 M1551P28 M1551P29 M1551P38 M1551P90 M1551P71
## <int> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 1 0 0 0 0 0 0 0 0 0
## 2 2 41 22 4 726 112492 2 413 2 1
## 3 3 67 65 12 13514 1 4 13314 70 5929
## 4 4 3229 13679 1832 951 113 2496 567 2428 2156
## 5 5 1200 92 3530 2008 0 183 2087 292 1058
## 6 6 219 1980 1200 499 1 781 214 2273 171
## 7 7 485 5123 755 4080 443 1278 2193 401 5320
## 8 8 840 7079 3760 22 0 2699 22 2870 32
## 9 9 40 82 91 881 1 449 1121 90 2283
## 10 10 11 79 277 2879 1 0 6811 14 243
## # ... with 47,796 more rows, and 1,689 more variables: M1551P12 <dbl>, M1551P84 <dbl>,
## # M1551P48 <dbl>, M1551P4 <dbl>, M1551P52 <dbl>, M1551P3 <dbl>, M1551P15 <dbl>, M1551P31 <dbl>,
## # M1551P75 <dbl>, M1551P88 <dbl>, …
这个大数据集,有47,806行(OTUs)和1.699列(1,698个sample和1个OTU ID)。如果你的计算机不能加载这个文件,不要担心,我们稍后将提供为剩下的workshop提供一个子集。
在这个数据集中, OTU的物种分类在一个不同的文件中。这些信息能够作为OTU表另外的列被包括,通常在其他数据集中。点击此处来下载物种分类表:
tax_data <- read_tsv("data/taxAssignments97.txt")
print(tax_data) # You can also enter `tax_data` to print it
结果为:
## # A tibble: 47,806 x 8
## `OTU ID` taxonomy Kingdom Phylum Class Order Family Confidence
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl>
## 1 OTU_1 Unassigned Unassig… Unassi… Unassig… Unassig… Unassign… 1.00
## 2 OTU_10 Root;k__Bacteria;p__Bacteroid… Bacteria Bacter… Sphingo… Sphingo… Sphingob… 1.00
## 3 OTU_100 Root;k__Bacteria;p__Cyanobact… Bacteria Cyanob… Chlorop… Chlorop… NA 1.00
## 4 OTU_1000 Root;k__Bacteria;p__Actinobac… Bacteria Actino… Actinob… Actinom… Actinosy… 0.670
## 5 OTU_10000 Unassigned Unassig… Unassi… Unassig… Unassig… Unassign… 1.00
## 6 OTU_10001 Root;k__Bacteria;p__Chlamydia… Bacteria Chlamy… Chlamyd… Chlamyd… Parachla… 1.00
## 7 OTU_10002 Root;k__Bacteria;p__Proteobac… Bacteria Proteo… Alphapr… NA NA 1.00
## 8 OTU_10003 Unassigned Unassig… Unassi… Unassig… Unassig… Unassign… 1.00
## 9 OTU_10004 Unassigned Unassig… Unassi… Unassig… Unassig… Unassign… 1.00
## 10 OTU_10005 Root;k__Bacteria;p__Cyanobact… Bacteria Cyanob… 4C0d-2 MLE1-12 NA 1.00
## # ... with 47,796 more rows
尽管这些数据相比大多数已经很好的格式化了,但是仍然有一些问题。“OTU ID”列的列名包含一个空格(因而出现了反引号),这使得用R尽心处理时更加恼火。更重要地,物种分类表中的OTU IDs带有前缀“OTU_”,但是OTU表格中的OTU IDs没有前缀,所以我们必须去掉前缀来使两张表相匹配。函数sub和gsub能够被用来进行文本查找和替换;sub只替换首个匹配项,而gsub则替换所有的匹配项。用nothing("")替换可以有效地搜索和删除。
tax_data$`OTU ID` <- sub(tax_data$`OTU ID`, # ` are needed because of the space
pattern = "OTU_", replacement = "")
print(tax_data)
结果为:
## # A tibble: 47,806 x 8
## `OTU ID` taxonomy Kingdom Phylum Class Order Family Confidence
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl>
## 1 1 Unassigned Unassig… Unassig… Unassig… Unassig… Unassign… 1.00
## 2 10 Root;k__Bacteria;p__Bacteroid… Bacteria Bactero… Sphingo… Sphingo… Sphingob… 1.00
## 3 100 Root;k__Bacteria;p__Cyanobact… Bacteria Cyanoba… Chlorop… Chlorop… NA 1.00
## 4 1000 Root;k__Bacteria;p__Actinobac… Bacteria Actinob… Actinob… Actinom… Actinosy… 0.670
## 5 10000 Unassigned Unassig… Unassig… Unassig… Unassig… Unassign… 1.00
## 6 10001 Root;k__Bacteria;p__Chlamydia… Bacteria Chlamyd… Chlamyd… Chlamyd… Parachla… 1.00
## 7 10002 Root;k__Bacteria;p__Proteobac… Bacteria Proteob… Alphapr… NA NA 1.00
## 8 10003 Unassigned Unassig… Unassig… Unassig… Unassig… Unassign… 1.00
## 9 10004 Unassigned Unassig… Unassig… Unassig… Unassig… Unassign… 1.00
## 10 10005 Root;k__Bacteria;p__Cyanobact… Bacteria Cyanoba… 4C0d-2 MLE1-12 NA 1.00
## # ... with 47,796 more rows
尽管我们能够使用分开的OTU和taxonomy表来进行分析,让我们结合它们来是事情更加简单吧!因为行是不同顺序的,我们需要基于OTU ID来combine(又称"join")它们。我们将使用dplyr package来做这个。
library(dplyr) # Loads the dplyr package so we can use `left_join`
tax_data$`OTU ID` <- as.character(tax_data$`OTU ID`) # Must be same type for join to work
otu_data$OTU_ID <- as.character(otu_data$OTU_ID) # Must be same type for join to work
otu_data <- left_join(otu_data, tax_data,
by = c("OTU_ID" = "OTU ID")) # identifies cols with shared IDs
print(otu_data)
结果为:
## # A tibble: 47,806 x 1,706
## OTU_ID M1024P1833 M1551P81 M1551P57 M1551P85 M1551P28 M1551P29 M1551P38 M1551P90 M1551P71
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 1 0. 0. 0. 0. 0. 0. 0. 0. 0.
## 2 2 41. 22. 4. 726. 112492. 2. 413. 2. 1.
## 3 3 67. 65. 12. 13514. 1. 4. 13314. 70. 5929.
## 4 4 3229. 13679. 1832. 951. 113. 2496. 567. 2428. 2156.
## 5 5 1200. 92. 3530. 2008. 0. 183. 2087. 292. 1058.
## 6 6 219. 1980. 1200. 499. 1. 781. 214. 2273. 171.
## 7 7 485. 5123. 755. 4080. 443. 1278. 2193. 401. 5320.
## 8 8 840. 7079. 3760. 22. 0. 2699. 22. 2870. 32.
## 9 9 40. 82. 91. 881. 1. 449. 1121. 90. 2283.
## 10 10 11. 79. 277. 2879. 1. 0. 6811. 14. 243.
## # ... with 47,796 more rows, and 1,696 more variables: M1551P12 <dbl>, M1551P84 <dbl>,
## # M1551P48 <dbl>, M1551P4 <dbl>, M1551P52 <dbl>, M1551P3 <dbl>, M1551P15 <dbl>, M1551P31 <dbl>,
## # M1551P75 <dbl>, M1551P88 <dbl>, …
有太多无法在输出展示的行,但是我们可以查看最后10个列名来查看它们的存在:
tail(colnames(otu_data), n = 10) # `tail` returns the last n elements
输出为:
## [1] "M1958P1043" "M1691P1526" "M1691P1557" "taxonomy" "Kingdom" "Phylum" "Class"
## [8] "Order" "Family" "Confidence"
接下来,让我们加载sample数据。点击这里来下载sample数据表。
sample_data <- read_tsv("data/SMD.txt",
col_types = "cccccccccccccccc") # each "c" means a column of "character"
print(sample_data) # You can also enter `sample_data` to print it
输出为:
## # A tibble: 1,698 x 16
## SampleID Name Plant_ID Type Experiment Cohort Harvested Age Site Treatment Line Genotype
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
## 1 M1024P17… R_026… R_026 root fieldBCMA 2008 2011 3 LTM field 26 ril
## 2 M1024P17… R_073… R_073 root fieldBCMA 2008 2011 3 LTM field 73 ril
## 3 M1024P17… R_088… R_088 root fieldBCMA 2009 2011 2 LTM field 88 ril
## 4 M1024P18… R_156… R_156 root fieldBCMA 2009 2011 2 LTM field 156 ril
## 5 M1955P804 1_A_1… 1_A_1 root ecoGH NA 2011 NA Duke MAHsoil par1… PAR
## 6 M1956P837 1_A_1… 1_A_12 root ecoGH NA 2011 NA Duke MAHsoil mah3… MAH
## 7 M1957P983 1_A_3… 1_A_3 root ecoGH NA 2011 NA Duke MAHsoil silL… SIL
## 8 M1956P845 1_A_4… 1_A_4 root ecoGH NA 2011 NA Duke MAHsoil par1… PAR
## 9 M1957P987 1_A_7… 1_A_7 root ecoGH NA 2011 NA Duke MAHsoil par9… PAR
## 10 M1957P923 1_A_8… 1_A_8 root ecoGH NA 2011 NA Duke MAHsoil mil5… MIL
## # ... with 1,688 more rows, and 4 more variables: Block <chr>, oldPlate <chr>, newPlate <chr>,
## # Analysis <chr>
注意现在otu_data中sample的数目等于sample_data中行的数目,otu_data的列名出现在"SampleID"列。这意味着sample_data$SampleID的内容可以被用来获取OTU表格子集列。
3.3 转化为taxmap的格式
尽管我们的数据现在已经在R中了,但它不是一种专门针对群落丰度数据的格式;所有R语言知道的是你有一些大表格。不同的针对群落(如:microbiome)分析的包需要不同形式或者classes的数据。在编程术语中,类(class)是存储数据和用来处理数据的一些函数的一种明确的方式。当你用这种方式格式化特定数据集时,我们称之为类的对象(object)或者实例(instance)。许多R包实现它们自己的类和函数来将数据转换为它们的格式,而有些包使用其他包中定义的类。在R中如何存储taxonomy分类丰度矩阵有多种选择(如,phyloseq对象),但是我们此处我们将使用taxa包中定义的类。taxa包的目标是提供一种通用的标准化方法,来操作分配给分类方法的任何类型的信息。Taxa包提供了一系列灵活的解析函数(parsers),能够读取几乎任何格式,给出正确的设置。你能够在如下地址读到更多用taxa来解析taxonomic数据的内容:https://github.com/ropensci/taxa#parsing-data。 我们增加到丰度矩阵的taxaonomic数据有如下的格式:
head(otu_data$taxonomy, 10)
输出如下:
## [1] "Unassigned"
## [2] "Root;k__Bacteria;p__Proteobacteria;c__Alphaproteobacteria;o__Rickettsiales;f__mitochondria"
## [3] "Root;k__Bacteria;p__Proteobacteria;c__Alphaproteobacteria;o__Sphingomonadales;f__Sphingomonadaceae"
## [4] "Root;k__Bacteria;p__Proteobacteria;c__Alphaproteobacteria;o__Rickettsiales;f__mitochondria"
## [5] "Root;k__Bacteria;p__Proteobacteria;c__Alphaproteobacteria;o__Rhizobiales;f__Rhizobiaceae"
## [6] "Root;k__Bacteria;p__Actinobacteria;c__Actinobacteria;o__Actinomycetales;f__Kineosporiaceae"
## [7] "Root;k__Bacteria;p__Cyanobacteria;c__Chloroplast;o__Streptophyta;f__"
## [8] "Root;k__Bacteria;p__Proteobacteria;c__Alphaproteobacteria;o__Rhizobiales;f__Bradyrhizobiaceae"
## [9] "Root;k__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Pseudomonadales;f__Pseudomonadaceae"
## [10] "Root;k__Bacteria;p__Bacteroidetes;c__Sphingobacteriia;o__Sphingobacteriales;f__Sphingobacteriaceae"
请注意,格式中有一些奇怪的方面可能会使解析变得很困难:
- 一些taxa拥有ranks(如,“k__Bacteria”),但是有些没有(如:”Unassigned" 和 “Root”)。
- 一些taxa拥有ranks,但是没有名字(如,“f__“)。
如果我们仅仅把ranks考虑为taxon name的一部分,那么它很容易解析:
library(taxa)
obj <- parse_tax_data(otu_data,
class_cols = "taxonomy", # The column in the input table
class_sep = ";") # What each taxon is seperated by
print(obj)
输出为:
## <Taxmap>
## 1558 taxa: aab. Unassigned, aac. Root ... chx. f__Methanospirillaceae, chy. f__
## 1558 edges: NA->aab, NA->aac, aac->aad, aac->aae ... bel->chw, ays->chx, bem->chy
## 1 data sets:
## tax_data:
## # A tibble: 47,806 x 1,707
## taxon_id OTU_ID M1024P1833 M1551P81 M1551P57 M1551P85 M1551P28 M1551P29 M1551P38
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 aab 1 0. 0. 0. 0. 0. 0. 0.
## 2 ben 2 41. 22. 4. 726. 112492. 2. 413.
## 3 beo 3 67. 65. 12. 13514. 1. 4. 13314.
## # ... with 4.78e+04 more rows, and 1,698 more variables: M1551P90 <dbl>,
## # M1551P71 <dbl>, M1551P12 <dbl>, M1551P84 <dbl>, M1551P48 <dbl>, M1551P4 <dbl>,
## # M1551P52 <dbl>, M1551P3 <dbl>, M1551P15 <dbl>, M1551P31 <dbl>, …
## 0 functions:
上面是taxmap对象的输出。第一行告诉我们OTUs被分配我1,558个独一无二的分类单元,列举出了它们的IDs和names。这些taxon IDs不存在与原始的数据集中,当转化为taxmap格式时自动产生。第二行描述了taxa跟树的其他节点联系。注意我们的原始数据在这个对象中的样子:
print(obj$data$tax_data)
输出如下:
## # A tibble: 47,806 x 1,707
## taxon_id OTU_ID M1024P1833 M1551P81 M1551P57 M1551P85 M1551P28 M1551P29 M1551P38 M1551P90
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 aab 1 0. 0. 0. 0. 0. 0. 0. 0.
## 2 ben 2 41. 22. 4. 726. 112492. 2. 413. 2.
## 3 beo 3 67. 65. 12. 13514. 1. 4. 13314. 70.
## 4 ben 4 3229. 13679. 1832. 951. 113. 2496. 567. 2428.
## 5 bep 5 1200. 92. 3530. 2008. 0. 183. 2087. 292.
## 6 beq 6 219. 1980. 1200. 499. 1. 781. 214. 2273.
## 7 ber 7 485. 5123. 755. 4080. 443. 1278. 2193. 401.
## 8 bes 8 840. 7079. 3760. 22. 0. 2699. 22. 2870.
## 9 bet 9 40. 82. 91. 881. 1. 449. 1121. 90.
## 10 beu 10 11. 79. 277. 2879. 1. 0. 6811. 14.
## # ... with 47,796 more rows, and 1,697 more variables: M1551P71 <dbl>, M1551P12 <dbl>,
## # M1551P84 <dbl>, M1551P48 <dbl>, M1551P4 <dbl>, M1551P52 <dbl>, M1551P3 <dbl>, M1551P15 <dbl>,
## # M1551P31 <dbl>, M1551P75 <dbl>, …
obj$data是任意的用户定义数据集的列表。这些数据集能够被命名为任何东西,可以是任何R对象,例如list, vector或者table。这与phyloseq对象不同,后者具有固定数量的预定义格式的数据集,因为taxa的重点是通常的taxonomy数据,而phyloseq的重点是特定的微生物组数据。注意,我们的数据集现在有一个"taxon_id"列,其将表格的行和taxonomy的taxa联系起来。这一列对于理解taxa的操纵函数如何处理这些数据集并且随后进行展示是非常重要的。
如果我们在解析时拆分rank的信息,我们可以使用正则表达式(regular expressions, 或称regex)来指定一个rank的每个taxon的部分或者名字的一部分。如果你对使用正则表达式不熟悉,刚开始可能很难理解,但是它是一项非常有用的技能,因此它值得学习。大多数正则表达式由一系列“匹配什么”以及“匹配多少次”组成。一个匹配taxon name模式的正则表达式是^[a-z]{0,1}_{0,2}.*$。这可能看起来比较吓人,但是我们可以将它分解为可以理解的部分:
^和$分别代表着文本的开头和末尾。如果没有这个,模式可能会匹配文本的一部分。- 方括号(如
[a-z])指定可以匹配的字符串范围。同样地,.意味着可以匹配任何字符串。 - 花括号的内容(如
{0,1})显示了此前模式能够匹配的数目。同样地,*意味着可以0次或者多次匹配。例如正则表达式的^[a-z]{0,1}部分意味着“在字符串的开头匹配从a到z的字母0次或者1次”。 - 任何不是特定正则表达式字符(如
[和.)的文本都可以匹配它自己,如_匹配文本中的_。为了匹配文本中像[的字符,你使用\\来对其进行换码(如\\[)。
整个正则表达式的意义是:
“从字符串的开头,(^)匹配任何从"a"到"z" ([a-z]) 的字符串0次或者1次({0,1}),接下来是下划线(_)出现0次至2次({0,2}), 接下来任何字符(.)出现0次或者多次,加下来是文本的结束($)。”
我们可以添加圆括号来指定模式的哪些部分组合在一起;这些在regex术语中称为捕获组(capture groups)。这些不会改变什么将被匹配;它们只是定义了模式的不同部分。在本例中,我们感兴趣的是分类单元级别(由([a-z]{0,1}匹配)和分类单元名称(由(.*匹配))。
taxa包中的parse_tax_data函数使用带有捕获组的正则表达式来分离我们想要的信息片段。对于正则表达式(又称为“regex”)中的每个捕获组,class_key选项将被赋予一个值,指定组是什么(例如,分类单元名称)。综上所述,我们可以这样解读数据:
obj <- parse_tax_data(otu_data,
class_cols = "taxonomy",
class_sep = ";",
class_regex = "^([a-z]{0,1})_{0,2}(.*)$",
class_key = c("tax_rank" = "taxon_rank", "name" = "taxon_name"))
print(obj)
输出为:
## <Taxmap>
## 1558 taxa: aab. Unassigned, aac. Root ... chx. Methanospirillaceae, chy.
## 1558 edges: NA->aab, NA->aac, aac->aad, aac->aae ... bel->chw, ays->chx, bem->chy
## 2 data sets:
## tax_data:
## # A tibble: 47,806 x 1,707
## taxon_id OTU_ID M1024P1833 M1551P81 M1551P57 M1551P85 M1551P28 M1551P29 M1551P38
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 aab 1 0. 0. 0. 0. 0. 0. 0.
## 2 ben 2 41. 22. 4. 726. 112492. 2. 413.
## 3 beo 3 67. 65. 12. 13514. 1. 4. 13314.
## # ... with 4.78e+04 more rows, and 1,698 more variables: M1551P90 <dbl>,
## # M1551P71 <dbl>, M1551P12 <dbl>, M1551P84 <dbl>, M1551P48 <dbl>, M1551P4 <dbl>,
## # M1551P52 <dbl>, M1551P3 <dbl>, M1551P15 <dbl>, M1551P31 <dbl>, …
## class_data:
## # A tibble: 216,417 x 5
## taxon_id input_index tax_rank name regex_match
## <chr> <int> <chr> <chr> <chr>
## 1 aab 1 "" Unassigned Unassigned
## 2 aac 2 "" Root Root
## 3 aad 2 k Bacteria k__Bacteria
## # ... with 2.164e+05 more rows
## 0 functions:
注意,分类单元名称不再具有rank信息:
head(taxon_names(obj)) #通过上面的输出可以看到,print(obj)的第一行就是taxa信息。
输出为:
## aab aac aad aae aaf
## "Unassigned" "Root" "Bacteria" "Archaea" "Proteobacteria"
## aag
## "Actinobacteria"
相反,rank信息(和任何其他捕获组内容)在一个单独的数据集中:
obj$data$class_data
输出为:
## # A tibble: 216,417 x 5
## taxon_id input_index tax_rank name regex_match
## <chr> <int> <chr> <chr> <chr>
## 1 aab 1 "" Unassigned Unassigned
## 2 aac 2 "" Root Root
## 3 aad 2 k Bacteria k__Bacteria
## 4 aaf 2 p Proteobacteria p__Proteobacteria
## 5 add 2 c Alphaproteobacteria c__Alphaproteobacteria
## 6 amg 2 o Rickettsiales o__Rickettsiales
## 7 ben 2 f mitochondria f__mitochondria
## 8 aac 3 "" Root Root
## 9 aad 3 k Bacteria k__Bacteria
## 10 aaf 3 p Proteobacteria p__Proteobacteria
## # ... with 216,407 more rows
但是,也可以使用taxon_rank函数获取rank:
head(taxon_ranks(obj))
输出为:
## aab aac aad aae aaf aag
## "" "" "k" "k" "p" "p"
因此我们并不真的需要“class_data”表,让我们删掉它:
obj$data$class_data <- NULL
我们还可以将“tax_data”表重命名为更有用的名称:
names(obj$data) <- "otu_counts"
print(obj)
输出结果为:
## <Taxmap>
## 1558 taxa: aab. Unassigned, aac. Root ... chx. Methanospirillaceae, chy.
## 1558 edges: NA->aab, NA->aac, aac->aad, aac->aae ... bel->chw, ays->chx, bem->chy
## 1 data sets:
## otu_counts:
## # A tibble: 47,806 x 1,707
## taxon_id OTU_ID M1024P1833 M1551P81 M1551P57 M1551P85 M1551P28 M1551P29 M1551P38
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 aab 1 0. 0. 0. 0. 0. 0. 0.
## 2 ben 2 41. 22. 4. 726. 112492. 2. 413.
## 3 beo 3 67. 65. 12. 13514. 1. 4. 13314.
## # ... with 4.78e+04 more rows, and 1,698 more variables: M1551P90 <dbl>,
## # M1551P71 <dbl>, M1551P12 <dbl>, M1551P84 <dbl>, M1551P48 <dbl>, M1551P4 <dbl>,
## # M1551P52 <dbl>, M1551P3 <dbl>, M1551P15 <dbl>, M1551P31 <dbl>, …
## 0 functions:
我们可以在obj$data中任意命名表或其他信息。obj$data是一个标准列表list,这意味着可以放入任何类型的任何数量的东西。
4.Exercises
4.1 Reading tabular data
- 查看文件example_data_1.tsv(点击下载)。
- 尝试使用基本R函数
read.table读取文件。您可能需要更改一些选项。输入?read.table以查看此函数的文档。提示:.tsv扩展名是什么意思?
注:.tsv扩展意味着文档是以tab键作为分隔符的。
?read.table
table1 = read.table("workshop\\import_data_into_R\\example_data_1.tsv", header = TRUE, sep = "\t")
print(table1)
- 现在尝试使用
readr包中的read_tsv函数读取相同的文件。键入?read_tsv查看此函数的文档。
library(readr)
?read_tsv
table2 = read_tsv("workshop\\import_data_into_R\\example_data_1.tsv")
print(table2)
- 比较这两个结果。他们有什么不同?尝试对每个结果使用str函数,查看它们的格式化细节。
str(table1)
str(table2)
使用基本R函数read.table读取文件获得的表格为data.drame,而使用readr包的read_tsv函数读取文件获得的表格为tibble(Classes ‘spec_tbl_df’, ‘tbl_df’, ‘tbl’ and ‘data.frame’)。
- 尝试更改这两个函数的输入参数,以便所有列都具有character类型(
str输出中的“chr”)。
table1 = read.table("workshop\\import_data_into_R\\example_data_1.tsv", header = TRUE, sep = "\t", colClasses="character")
table2 = read_tsv("workshop\\import_data_into_R\\example_data_1.tsv", col_types = "cccc")
4.2 Reading taxonomic data
下面的练习中我们将把前面练习得到的表格转化为taxmap对象。
my_data <- read_tsv("workshop\\import_data_into_R\\example_data_1.tsv")
print(my_data)
- 查看
parse_tax_data和lookup_tax_data的文档。文档底部的示例应该很有帮助。
library(taxa)
?parse_tax_data #读取物种分类以及表、列表、向量中的相关数据,存储到taxa::taxmap()对象。
?lookup_tax_data #从表、列表或向量中出现的NCBI sequence ID、Taxonomy ID或Taxonomy name查找分类数据。
- 尝试使用
parse_tax_data将表转换为taxmap对象,使用my_taxonomy列获取分类信息。您应该得到一个以“哺乳类”为根的分类法。输出中包含的表与输入表有何不同?
parse_tax_data(my_data, class_cols = "my_taxonomy", class_sep = ", ")
- 尝试使用
lookup_tax_data将表转换为taxmap对象,使用ncbi_seq_id列查找与这些Genbank登录号关联的分类法。注意:这需要网络连接。
lookup_tax_data(my_data, type = "seq_id", column = "ncbi_seq_id", database = "ncbi")
- 尝试使用lookup_tax_data将表转换为taxmap对象,使用itis_taxon_id列从集成分类信息系统(Integrated taxonomic Information System, ITIS)查找这些taxon IDs的taxonomic分类。注意:这需要网络连接。
lookup_tax_data(my_data, type = "taxon_id", column = "itis_taxon_id", database = "itis")
- 比较三种分类信息来源的结果。不同的是什么?相同的是什么?
- taxa的数量和名字不同,因为每个资源有不同的taxonomy。
- taxa IDs不同。当在线访问数据库时,taxon IDs来自访问的数据库。
- 存储在“tax_data”的输入数据是相同的,除了taxon ID列。
4.3 Reading taxonomic data from complex formats
有时候,分类数据可以嵌入到复杂的文本中,比如FASTA headers。查看example_data_2.fa(点击下载)文件,包含两个分类信息来源的headers:
- The Genbank accession number
- The taxonomic classfication
- 用
ape包的read.FASTA函数读取FASTA文件example_data_2.fa,将结果存储到一个变量。
library(ape)
seqs <- read.FASTA("workshop\\import_data_into_R\\example_data_2.fa")
print(seqs)
ape包:Analyses of Phylogenetics and Evolution。ape提供了读、写、操作、分析、仿真进化树、DNA序列、计算DNA距离、翻译成氨基酸序列、基于距离的方法来评估树以及各种比较分析和多样化分析的方法。
- 使用
names函数获取header信息,并将其存储在另一个变量中。
headers <- names(seqs)
print(headers)
-
通过键入
?extract_tax_data查看taxa包中extract_tax_data的文档。
答:extract_tax_data函数将字符向量的taxonomy信息转化为一个taxmap()对象。 -
使用
extract_tax_data函数和NCBI accession number将header转换为taxmap对象。分类可以忽略(即不给捕获组和键值)或存储为“info”。
extract_tax_data(headers,
key = c(my_acc_no = "seq_id", my_class = "info"),
regex = "^(.+)::(.+)$")
- 现在使用classfication而不是accession number。登录号可以忽略或存储为“info”。现在考虑将rank作为taxon name的一部分。
extract_tax_data(headers,
key = c(my_acc_no = "info", my_class = "class"),
regex = "^(.+)::(.+)$",
class_sep = ";")
- 现在尝试从taxon name中分离出rank信息。您将需要使用class_key和class_regex选项。这些列可以存储为“info”或“taxon_rank”。使用“taxon_rank”允许使用taxon_rank函数访问这些rank。
extract_tax_data(headers,
key = c(acc_no = "info", my_class = "class"),
regex = "^(.+)::(.+)$",
class_sep = ";",
class_key = c(tax_rank = "info", name = "taxon_name"),
class_regex = "^(.+)_(.+)$")
- 有时您可以选择使用多个taxonomic信息源,如本练习中所示。使用嵌入式分类信息而不是Genbank accession number或taxon ID有什么好处?
答:使用包含的分类不需要访问在线数据库,更加快捷和可依赖。
References
1.Installing required software and data
Dixon, Philip. 2003. “VEGAN, a Package of R Functions for Community Ecology.” Journal of Vegetation Science 14 (6). BioOne: 927–30.
Foster, Zachary SL, Scott Chamberlain, and Niklaus J Grünwald. 2018. “Taxa: An R Package Implementing Data Standards and Methods for Taxonomic Data.” F1000Research 7 (272). doi:10.12688/f1000research.14013.1.
Foster, Zachary SL, Thomas J Sharpton, and Niklaus J Grünwald. 2017. “Metacoder: An R Package for Visualization and Manipulation of Community Taxonomic Diversity Data.” PLoS Computational Biology 13 (2). Public Library of Science: e1005404. https://doi.org/10.1371/journal.pcbi.1005404.
McMurdie, Paul J, and Susan Holmes. 2013. “Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data.” PloS One 8 (4). Public Library of Science: e61217. https://doi.org/10.1371/journal.pone.0061217.
Wickham, Hadley. 2009. Ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York. http://ggplot2.org.
Wickham, Hadley, and Romain Francois. 2015. “Dplyr: A Grammar of Data Manipulation.” R Package Version 0.4 3.
2.Datasets used in this workshop
Wagner, Maggie R, Derek S Lundberg, G Tijana, Susannah G Tringe, Jeffery L Dangl, and Thomas Mitchell-Olds. 2016. “Host Genotype and Age Shape the Leaf and Root Microbiomes of a Wild Perennial Plant.” Nature Communications 7. Nature Publishing Group: 12151. doi:10.1038/ncomms12151.
3.Getting data into R
Wagner, Maggie R, Derek S Lundberg, G Tijana, Susannah G Tringe, Jeffery L Dangl, and Thomas Mitchell-Olds. 2016. “Host Genotype and Age Shape the Leaf and Root Microbiomes of a Wild Perennial Plant.” Nature Communications 7. Nature Publishing Group: 12151. doi:10.1038/ncomms12151.
Zeeberg, Barry R, Joseph Riss, David W Kane, Kimberly J Bussey, Edward Uchio, W Marston Linehan, J Carl Barrett, and John N Weinstein. 2004. “Mistaken Identifiers: Gene Name Errors Can Be Introduced Inadvertently When Using Excel in Bioinformatics.” BMC Bioinformatics 5 (1). BioMed Central: 80. https://doi.org/10.1186/1471-2105-5-80.
智能推荐
Docker 快速上手学习入门教程_docker菜鸟教程-程序员宅基地
文章浏览阅读2.5w次,点赞6次,收藏50次。官方解释是,docker 容器是机器上的沙盒进程,它与主机上的所有其他进程隔离。所以容器只是操作系统中被隔离开来的一个进程,所谓的容器化,其实也只是对操作系统进行欺骗的一种语法糖。_docker菜鸟教程
电脑技巧:Windows系统原版纯净软件必备的两个网站_msdn我告诉你-程序员宅基地
文章浏览阅读5.7k次,点赞3次,收藏14次。该如何避免的,今天小编给大家推荐两个下载Windows系统官方软件的资源网站,可以杜绝软件捆绑等行为。该站提供了丰富的Windows官方技术资源,比较重要的有MSDN技术资源文档库、官方工具和资源、应用程序、开发人员工具(Visual Studio 、SQLServer等等)、系统镜像、设计人员工具等。总的来说,这两个都是非常优秀的Windows系统镜像资源站,提供了丰富的Windows系统镜像资源,并且保证了资源的纯净和安全性,有需要的朋友可以去了解一下。这个非常实用的资源网站的创建者是国内的一个网友。_msdn我告诉你
vue2封装对话框el-dialog组件_<el-dialog 封装成组件 vue2-程序员宅基地
文章浏览阅读1.2k次。vue2封装对话框el-dialog组件_
MFC 文本框换行_c++ mfc同一框内输入二行怎么换行-程序员宅基地
文章浏览阅读4.7k次,点赞5次,收藏6次。MFC 文本框换行 标签: it mfc 文本框1.将Multiline属性设置为True2.换行是使用"\r\n" (宽字符串为L"\r\n")3.如果需要编辑并且按Enter键换行,还要将 Want Return 设置为 True4.如果需要垂直滚动条的话将Vertical Scroll属性设置为True,需要水平滚动条的话将Horizontal Scroll属性设_c++ mfc同一框内输入二行怎么换行
redis-desktop-manager无法连接redis-server的解决方法_redis-server doesn't support auth command or ismis-程序员宅基地
文章浏览阅读832次。检查Linux是否是否开启所需端口,默认为6379,若未打开,将其开启:以root用户执行iptables -I INPUT -p tcp --dport 6379 -j ACCEPT如果还是未能解决,修改redis.conf,修改主机地址:bind 192.168.85.**;然后使用该配置文件,重新启动Redis服务./redis-server redis.conf..._redis-server doesn't support auth command or ismisconfigured. try
实验四 数据选择器及其应用-程序员宅基地
文章浏览阅读4.9k次。济大数电实验报告_数据选择器及其应用
随便推点
灰色预测模型matlab_MATLAB实战|基于灰色预测河南省社会消费品零售总额预测-程序员宅基地
文章浏览阅读236次。1研究内容消费在生产中占据十分重要的地位,是生产的最终目的和动力,是保持省内经济稳定快速发展的核心要素。预测河南省社会消费品零售总额,是进行宏观经济调控和消费体制改变创新的基础,是河南省内人民对美好的全面和谐社会的追求的要求,保持河南省经济稳定和可持续发展具有重要意义。本文建立灰色预测模型,利用MATLAB软件,预测出2019年~2023年河南省社会消费品零售总额预测值分别为21881...._灰色预测模型用什么软件
log4qt-程序员宅基地
文章浏览阅读1.2k次。12.4-在Qt中使用Log4Qt输出Log文件,看这一篇就足够了一、为啥要使用第三方Log库,而不用平台自带的Log库二、Log4j系列库的功能介绍与基本概念三、Log4Qt库的基本介绍四、将Log4qt组装成为一个单独模块五、使用配置文件的方式配置Log4Qt六、使用代码的方式配置Log4Qt七、在Qt工程中引入Log4Qt库模块的方法八、获取示例中的源代码一、为啥要使用第三方Log库,而不用平台自带的Log库首先要说明的是,在平时开发和调试中开发平台自带的“打印输出”已经足够了。但_log4qt
100种思维模型之全局观思维模型-67_计算机中对于全局观的-程序员宅基地
文章浏览阅读786次。全局观思维模型,一个教我们由点到线,由线到面,再由面到体,不断的放大格局去思考问题的思维模型。_计算机中对于全局观的
线程间控制之CountDownLatch和CyclicBarrier使用介绍_countdownluach于cyclicbarrier的用法-程序员宅基地
文章浏览阅读330次。一、CountDownLatch介绍CountDownLatch采用减法计算;是一个同步辅助工具类和CyclicBarrier类功能类似,允许一个或多个线程等待,直到在其他线程中执行的一组操作完成。二、CountDownLatch俩种应用场景: 场景一:所有线程在等待开始信号(startSignal.await()),主流程发出开始信号通知,既执行startSignal.countDown()方法后;所有线程才开始执行;每个线程执行完发出做完信号,既执行do..._countdownluach于cyclicbarrier的用法
自动化监控系统Prometheus&Grafana_-自动化监控系统prometheus&grafana实战-程序员宅基地
文章浏览阅读508次。Prometheus 算是一个全能型选手,原生支持容器监控,当然监控传统应用也不是吃干饭的,所以就是容器和非容器他都支持,所有的监控系统都具备这个流程,_-自动化监控系统prometheus&grafana实战
React 组件封装之 Search 搜索_react search-程序员宅基地
文章浏览阅读4.7k次。输入关键字,可以通过键盘的搜索按钮完成搜索功能。_react search